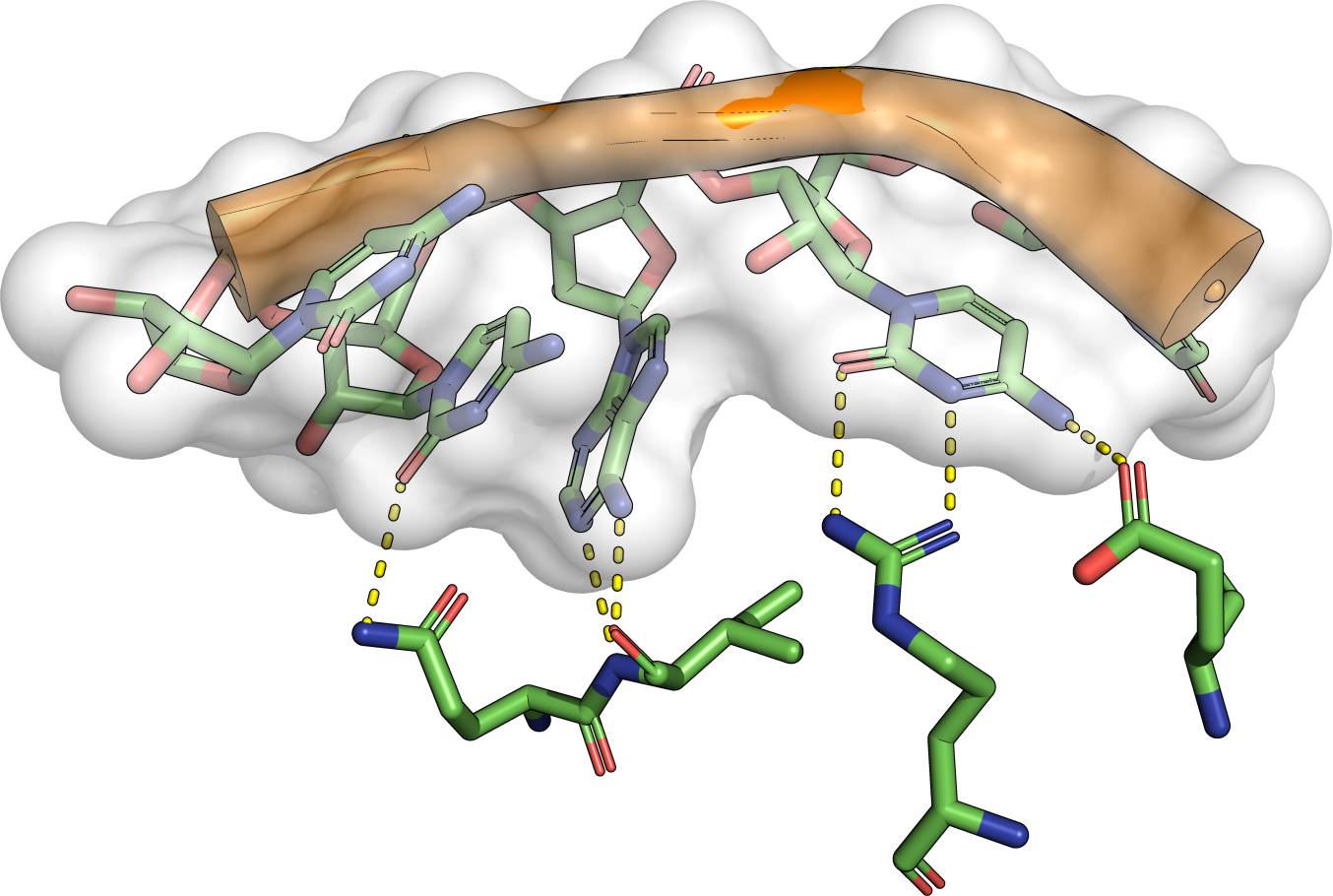
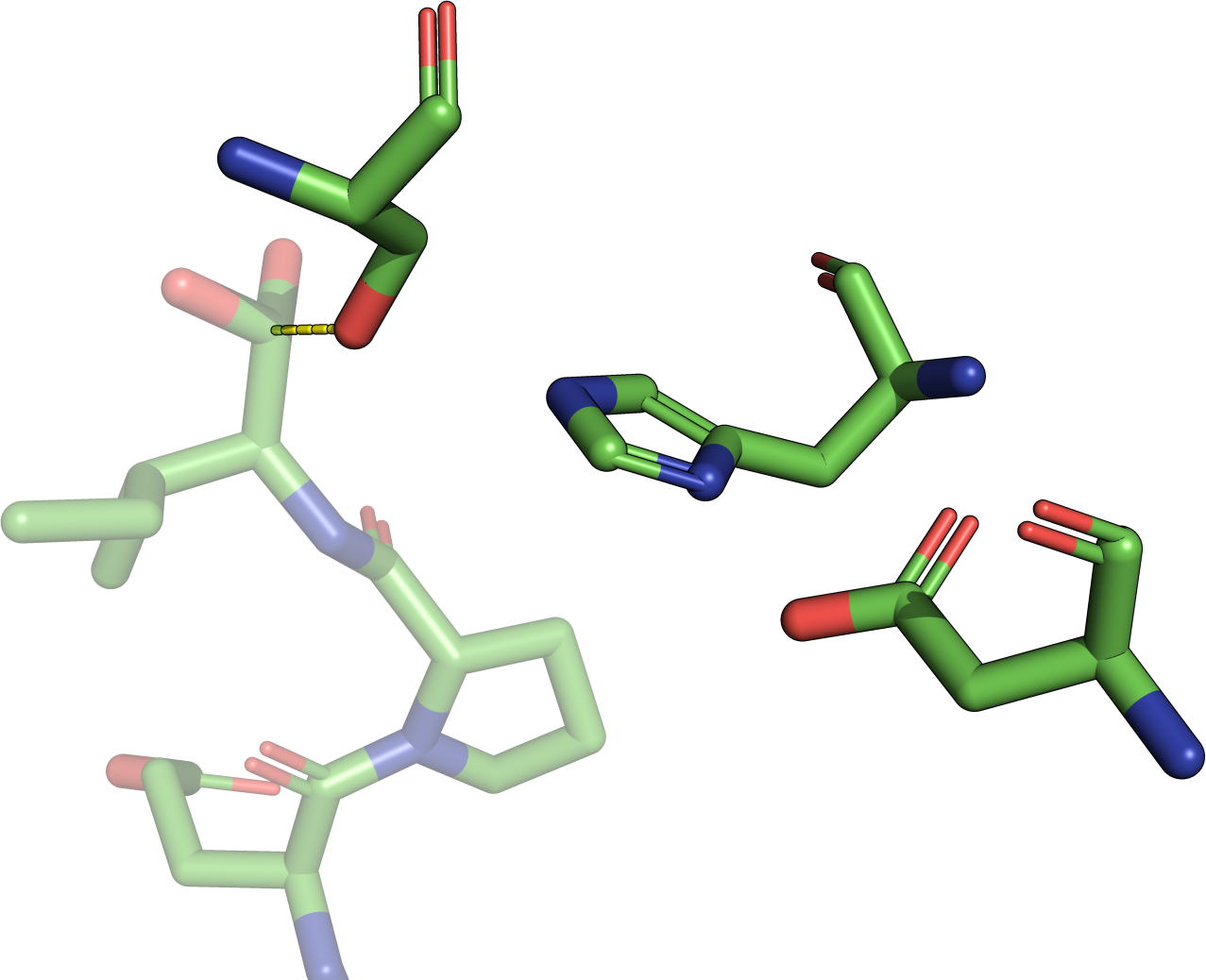
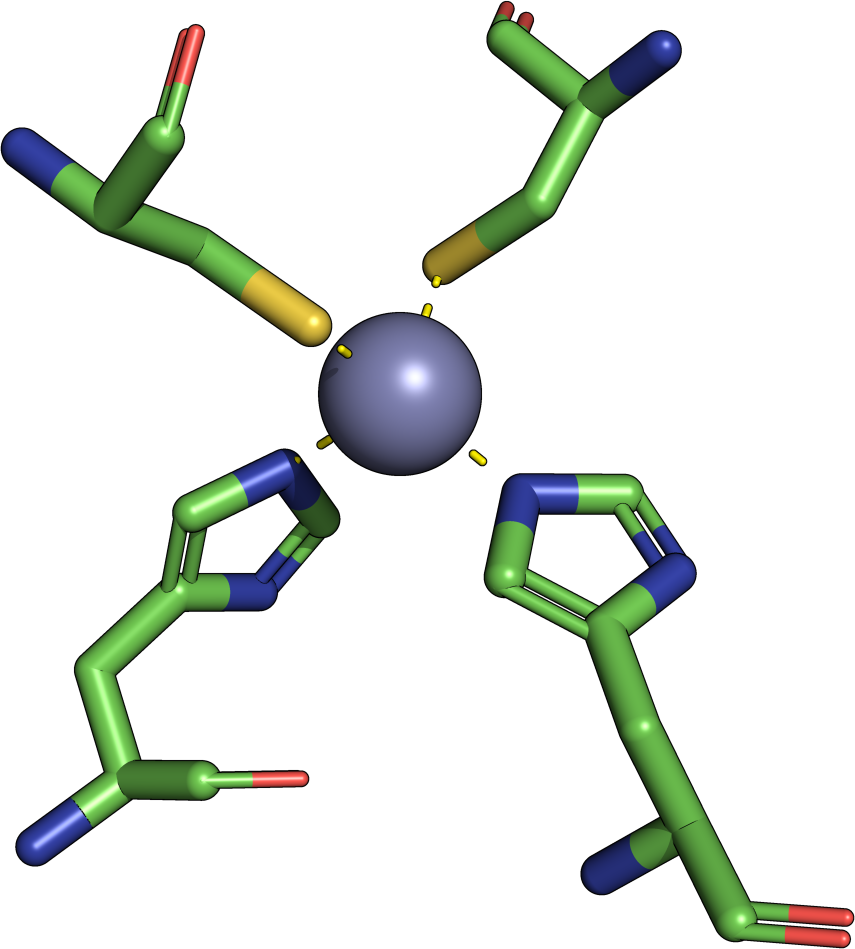
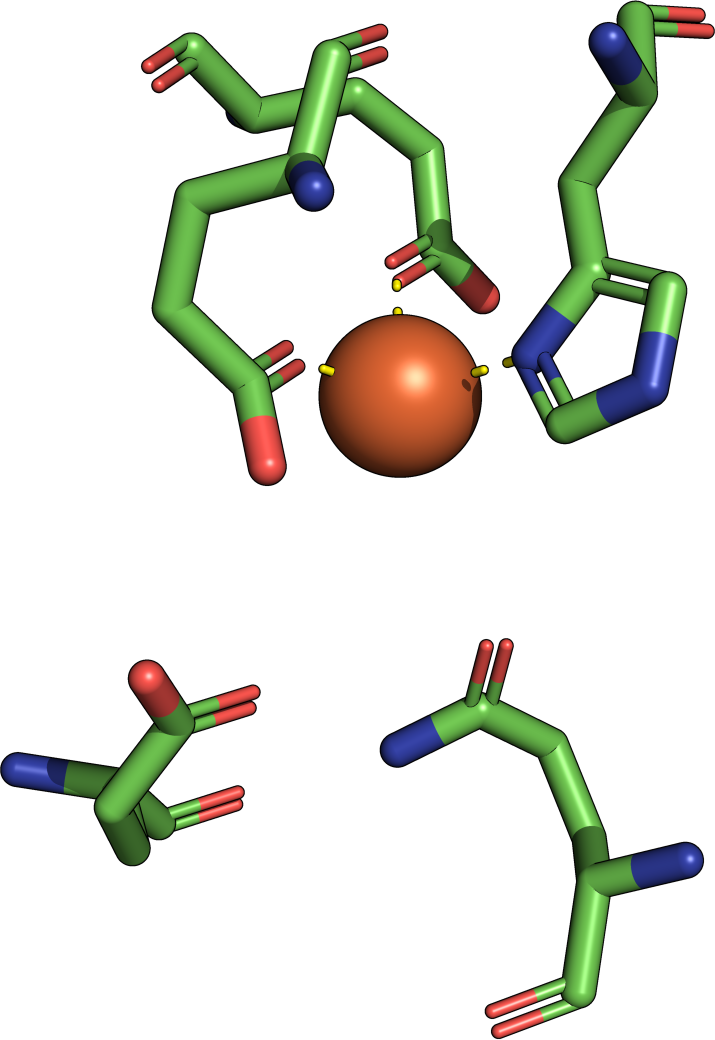
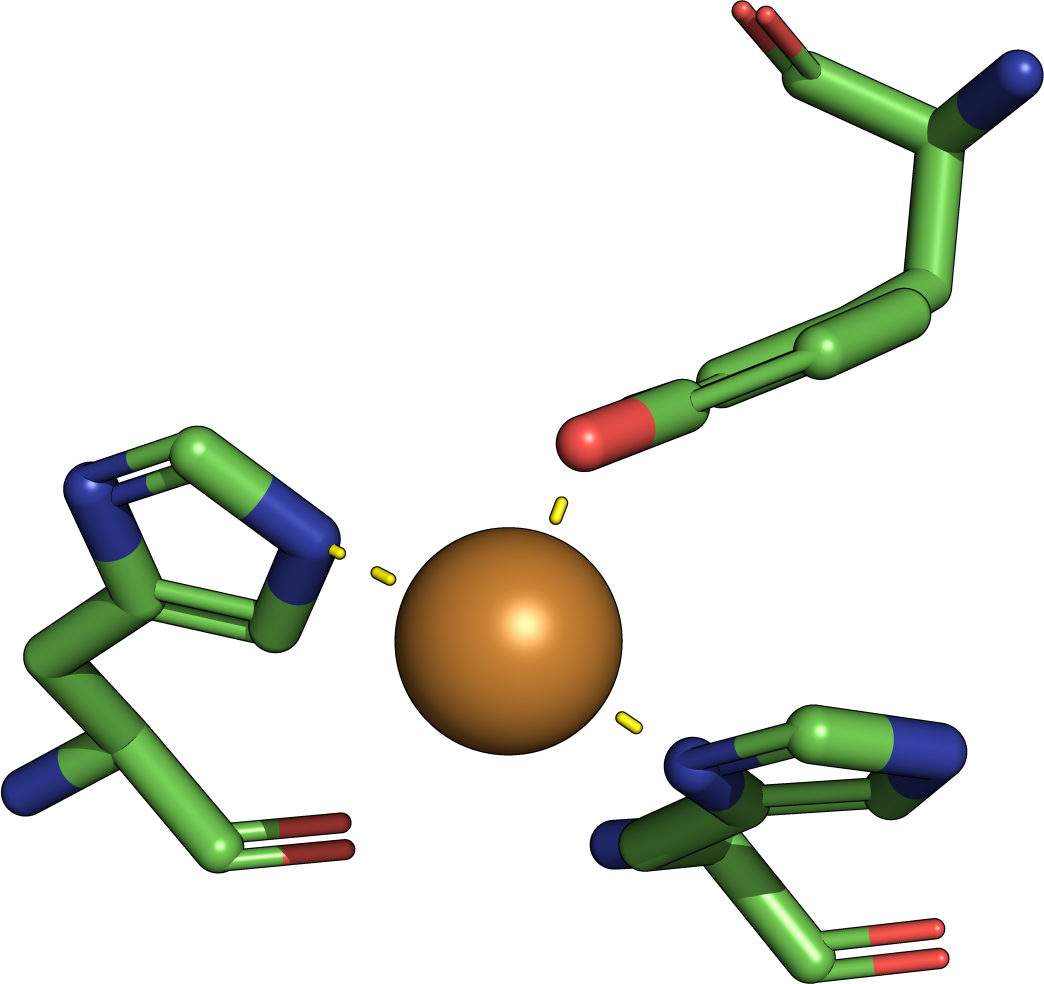
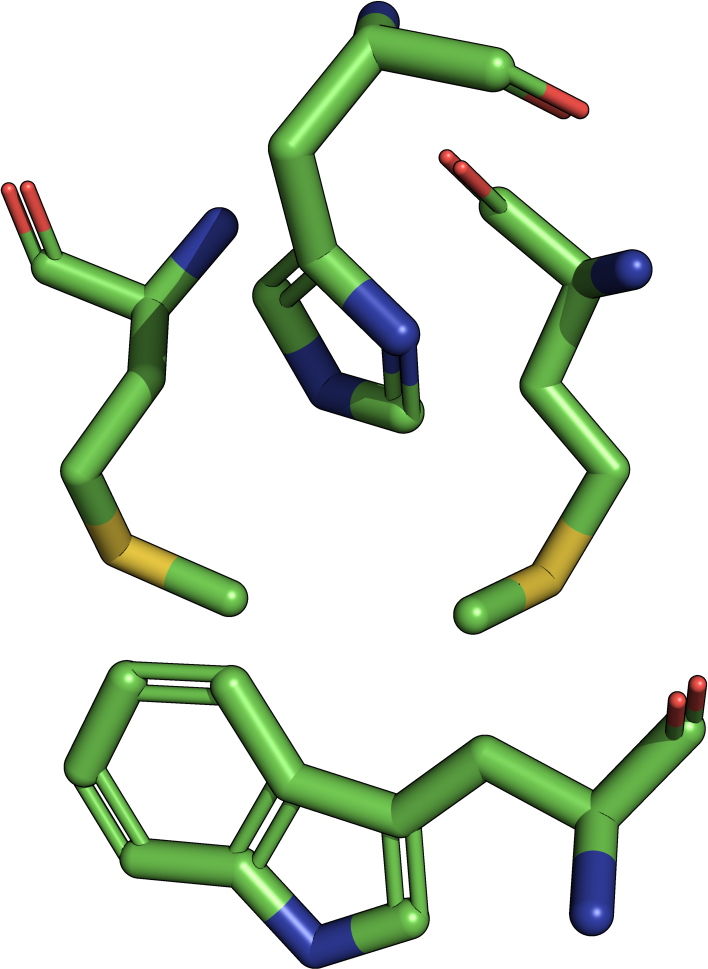
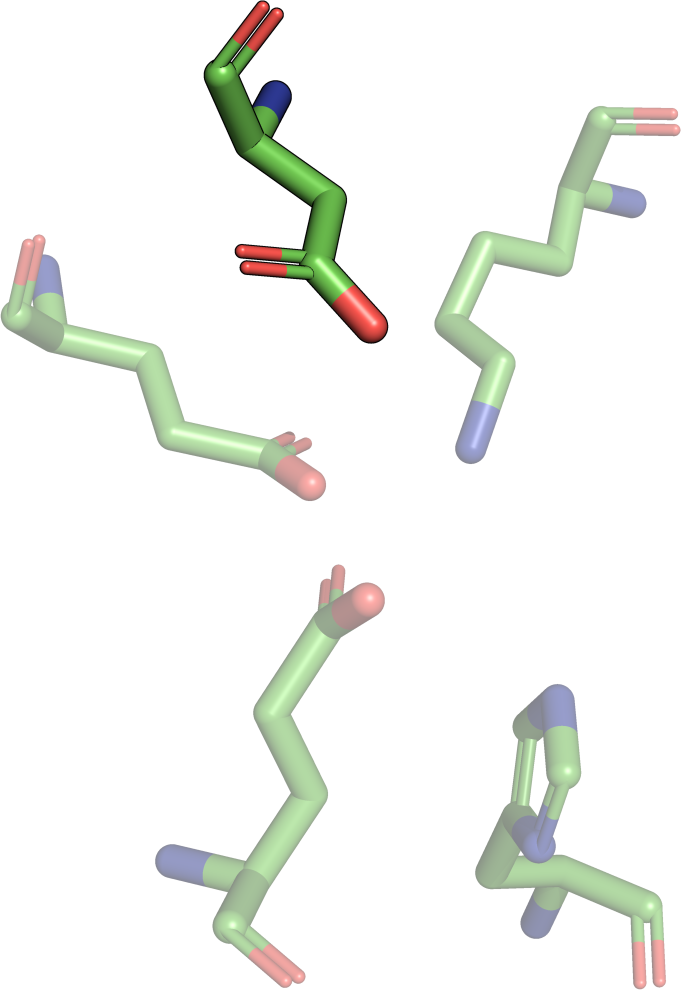
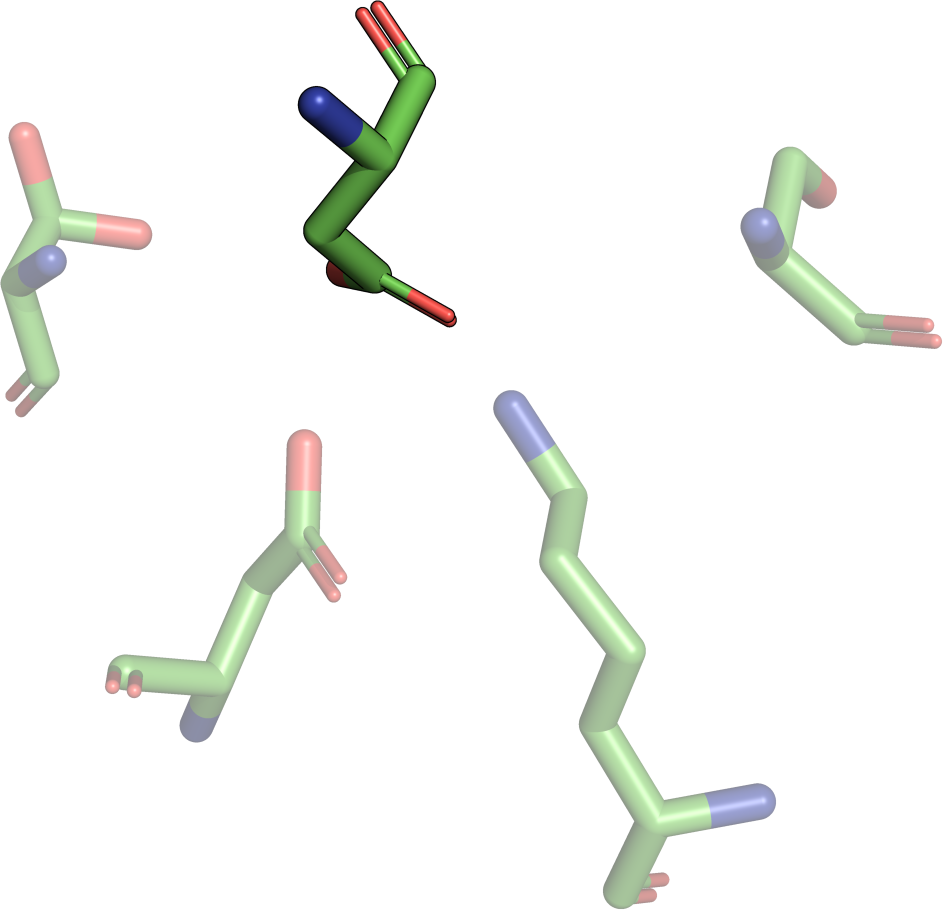
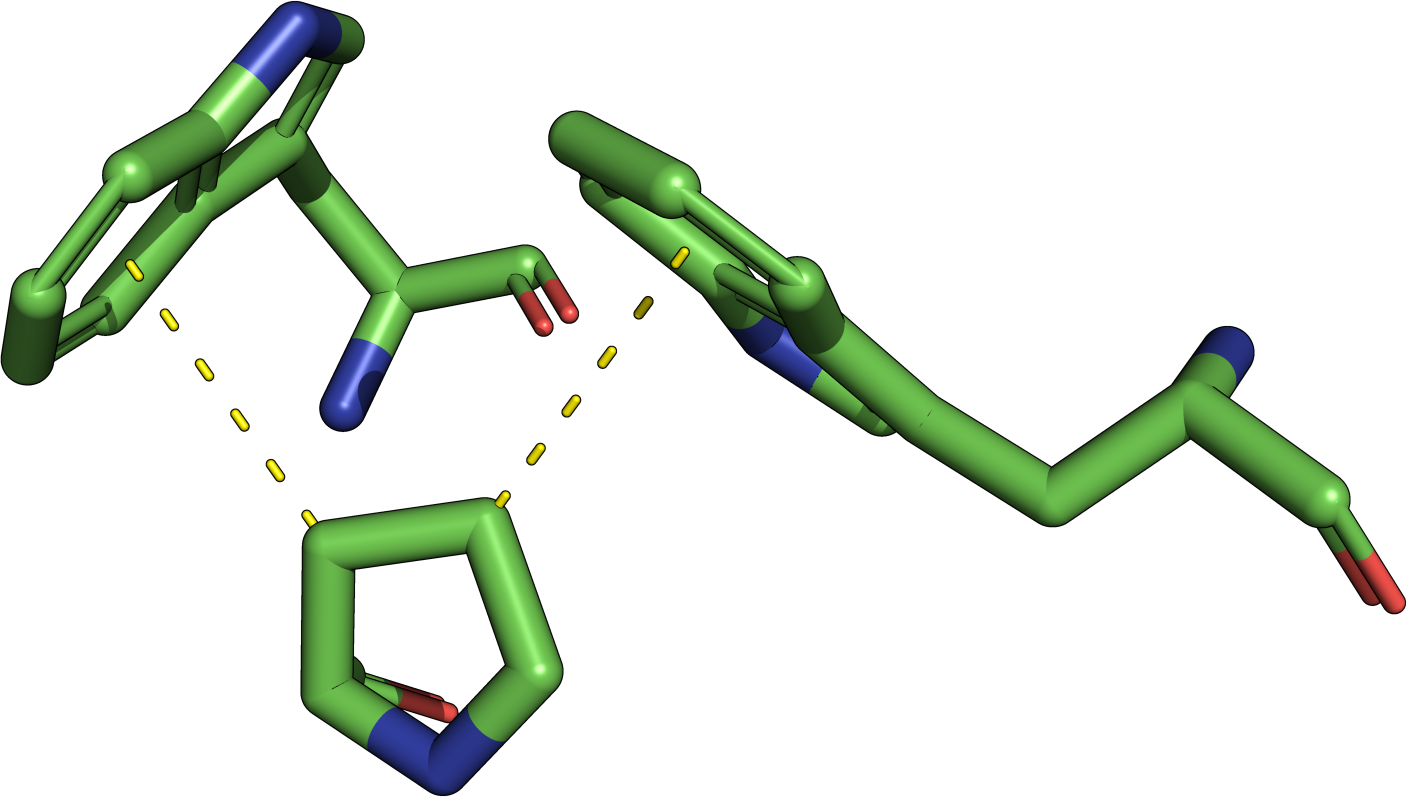
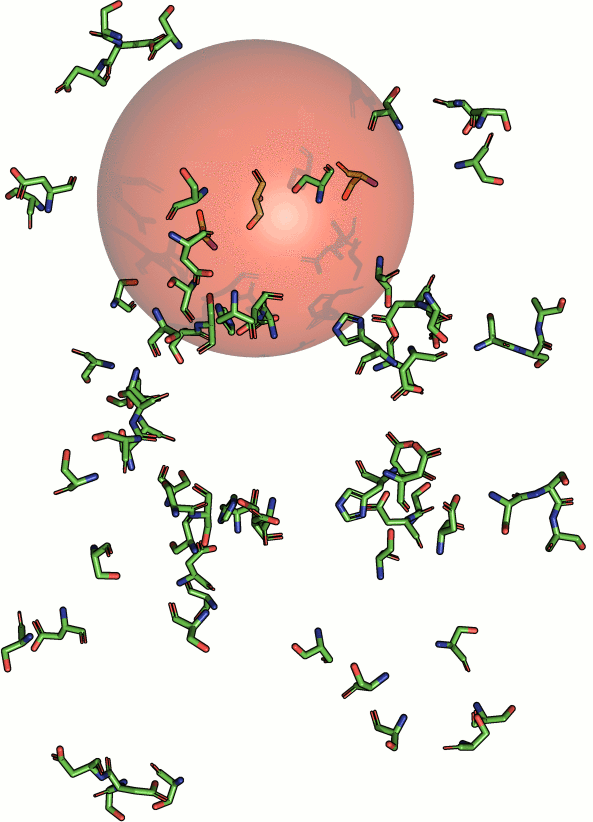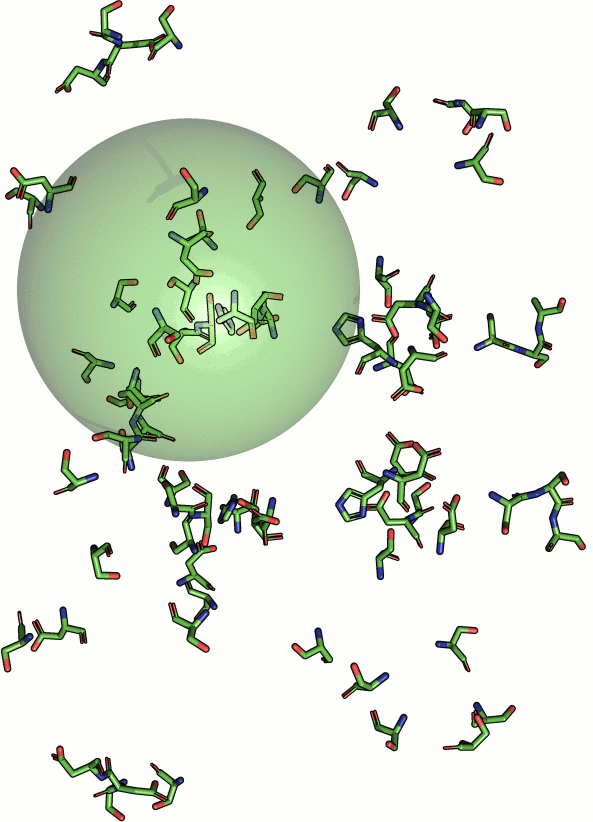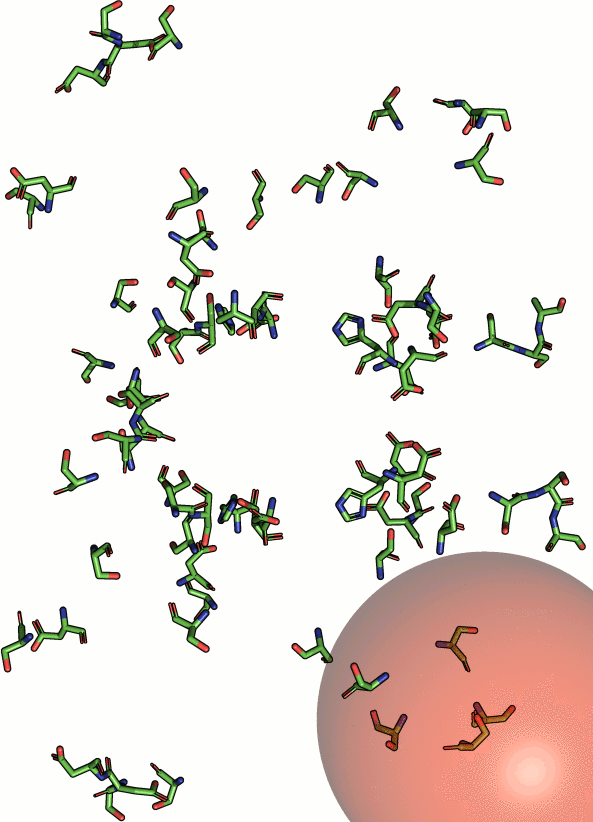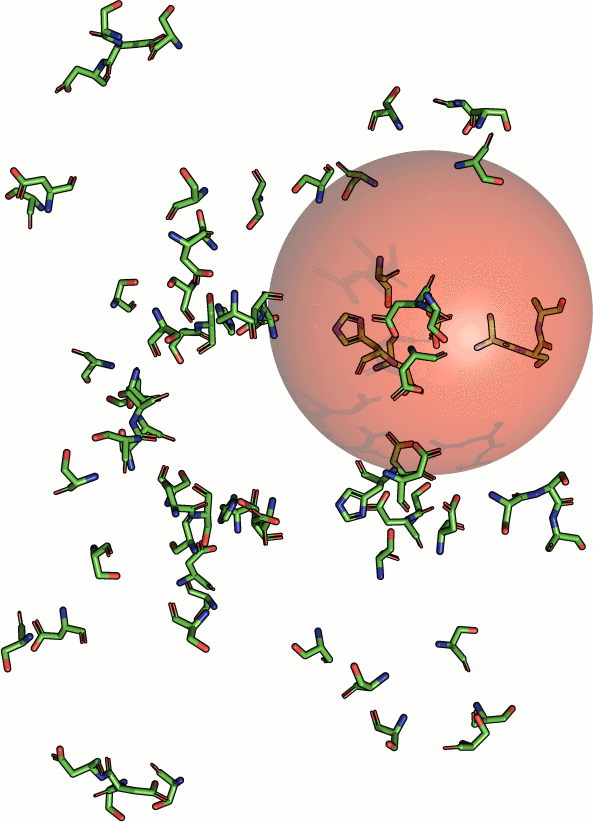
|
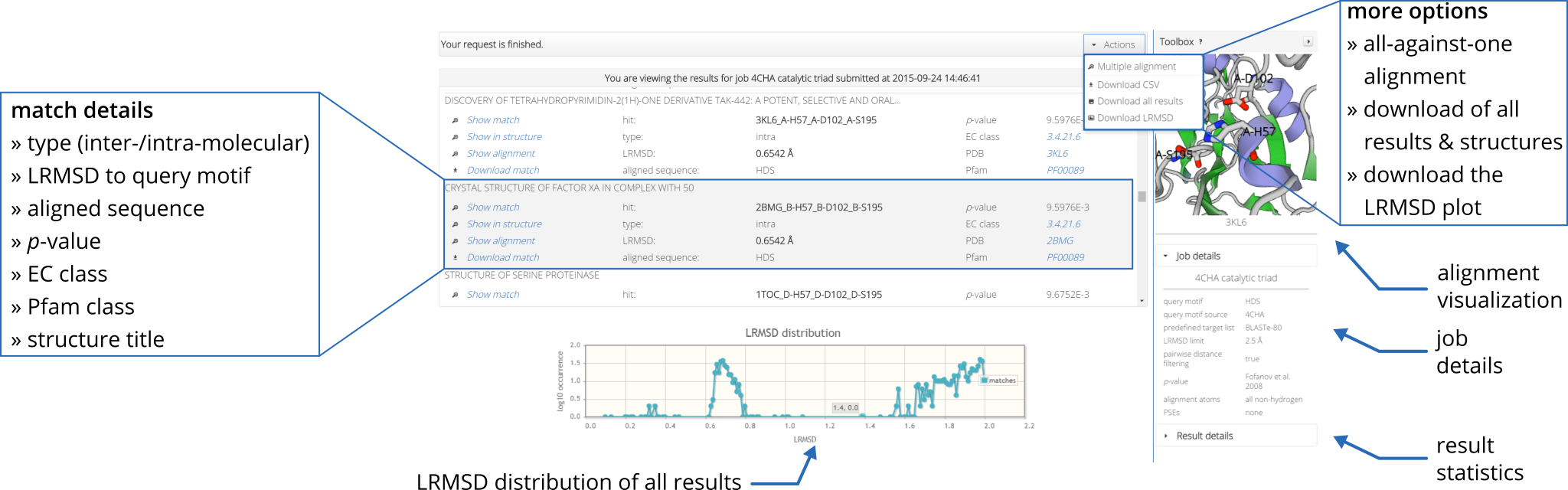
Beside geometric similarity matches are rated according their
statistical significance. Every reported match to a query motif
is labeled with a p-value such that the user can assess
true positives intuitively. However, a significant p-value
does not necessarily indicate a true positive match
because the underlying models are based solely on the
distribution of RMSD values. Hints on how to understand and
interpret the p-value are given in the
FAQ section.
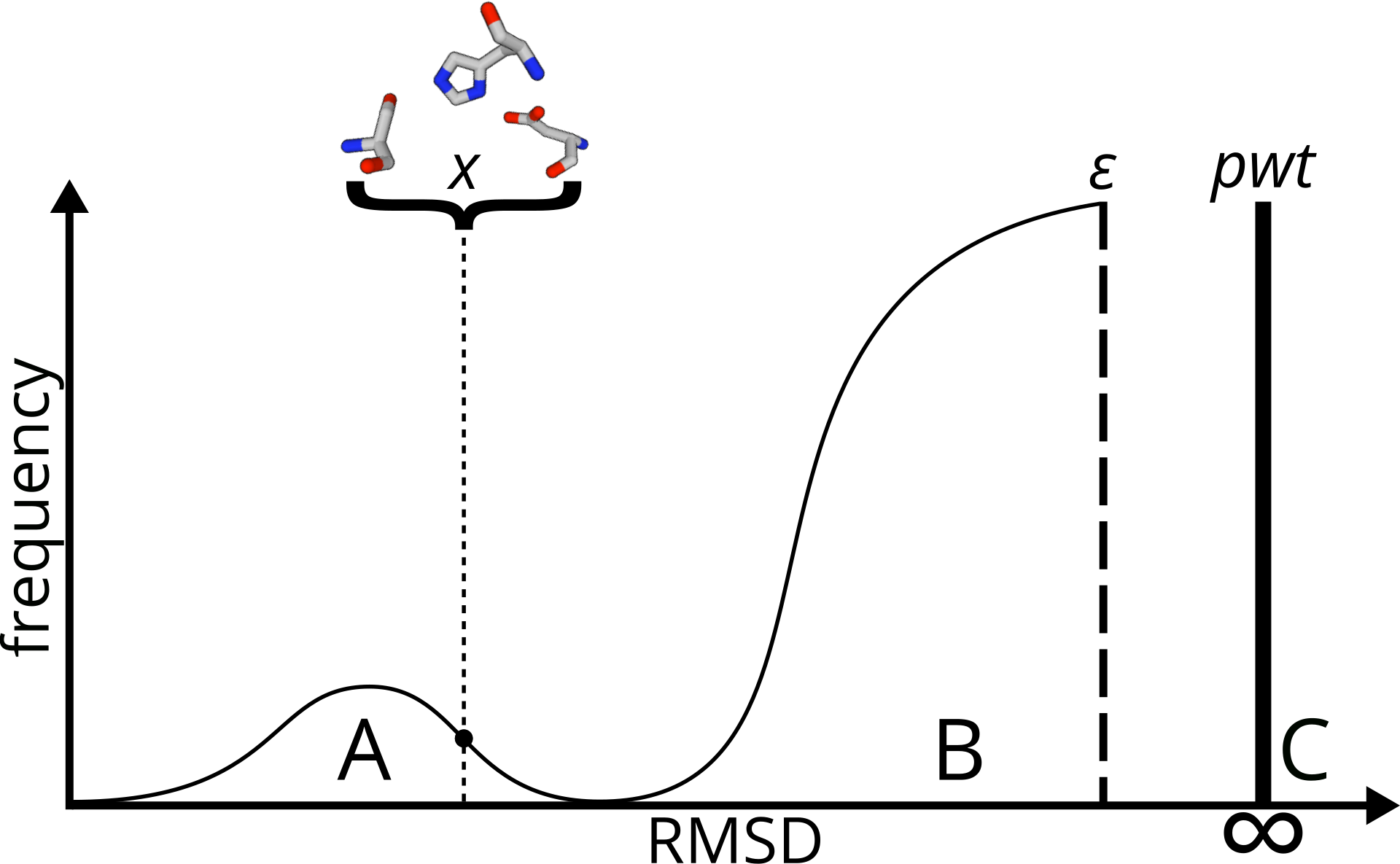
No biological features are represented by those models. To
estimate statistical significance, Fit3D implements two
statistical models:
- kernel density based model, corrected with a maximum
likelihood estimated point-weight (according to Fofanov et al.
2008 )
- empirically derived model according to Stark et al. 2003
| |