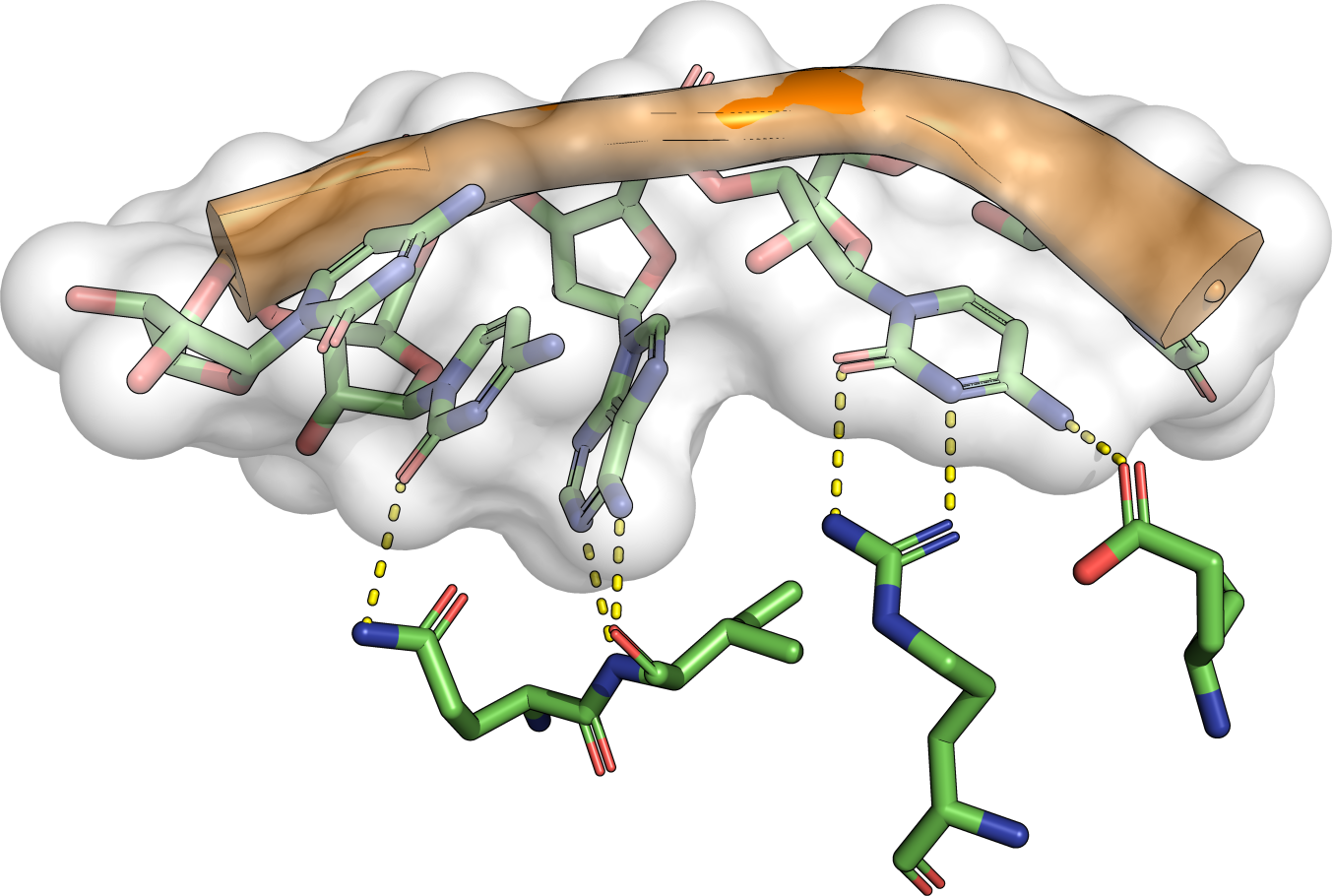
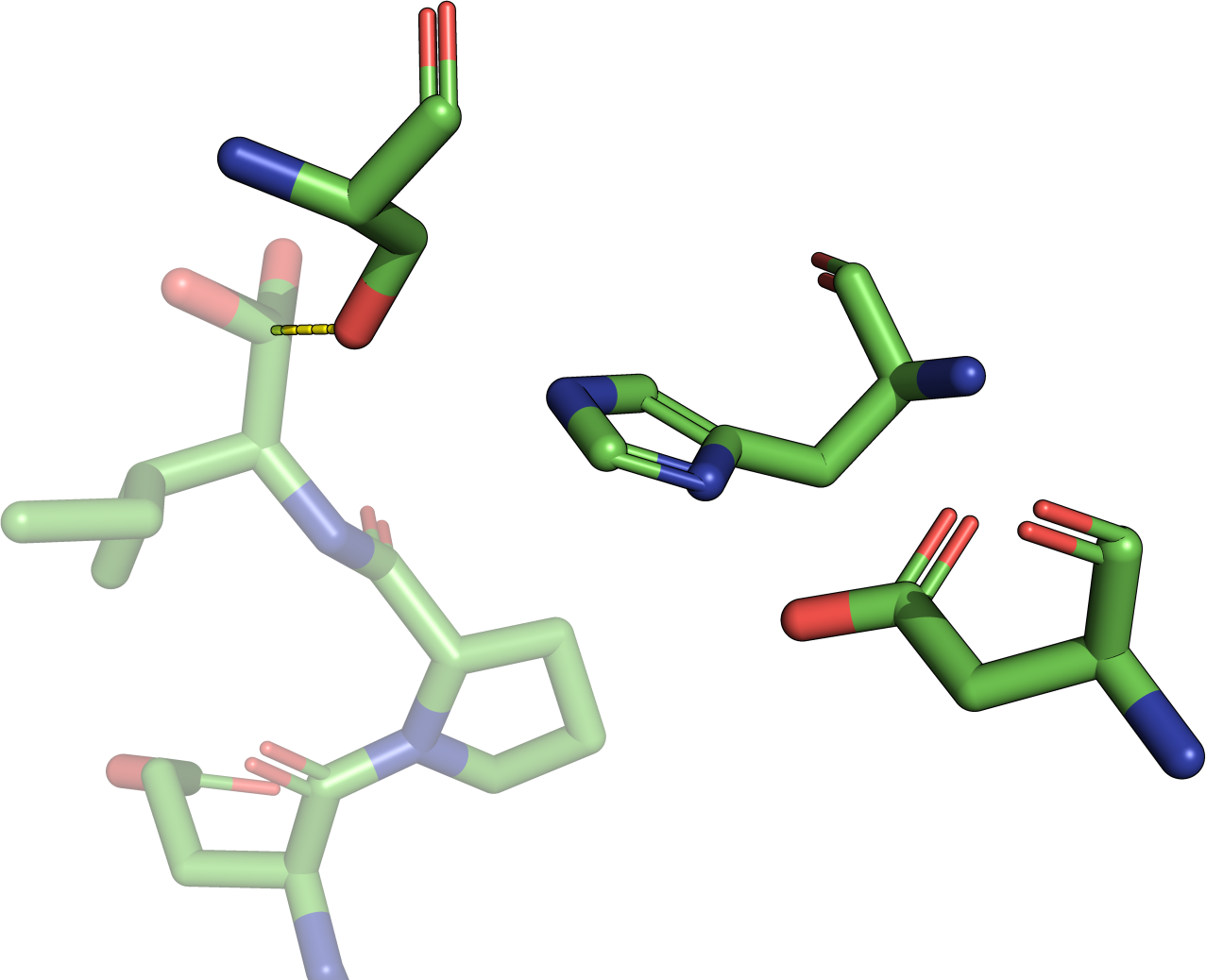
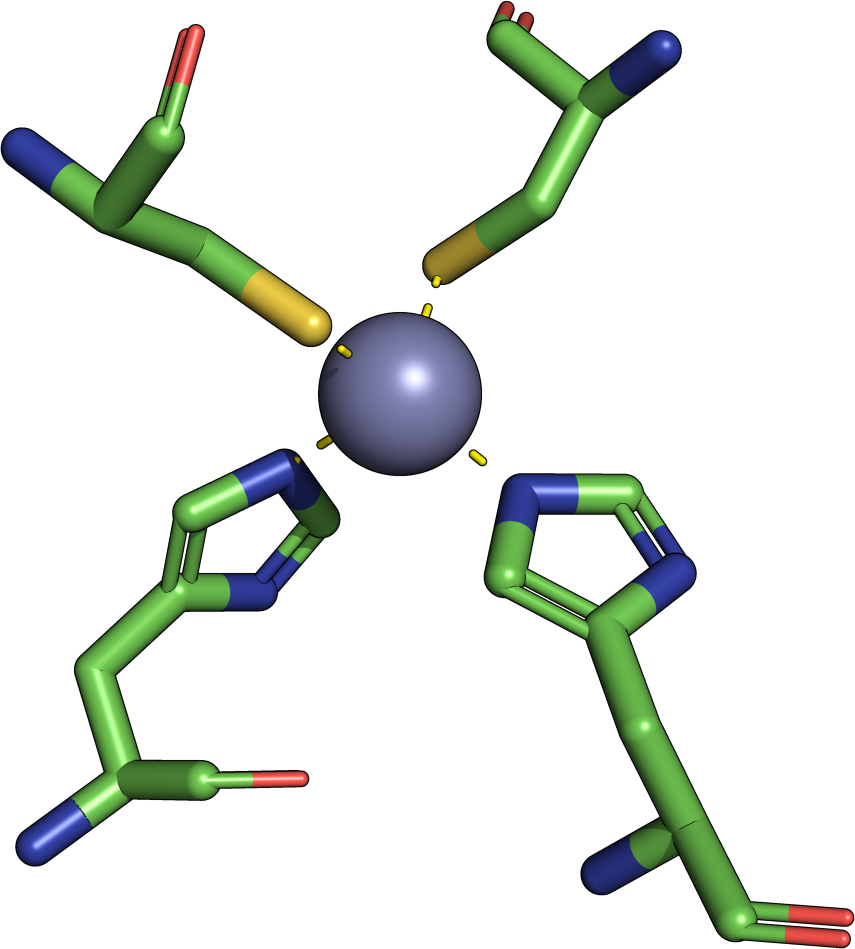
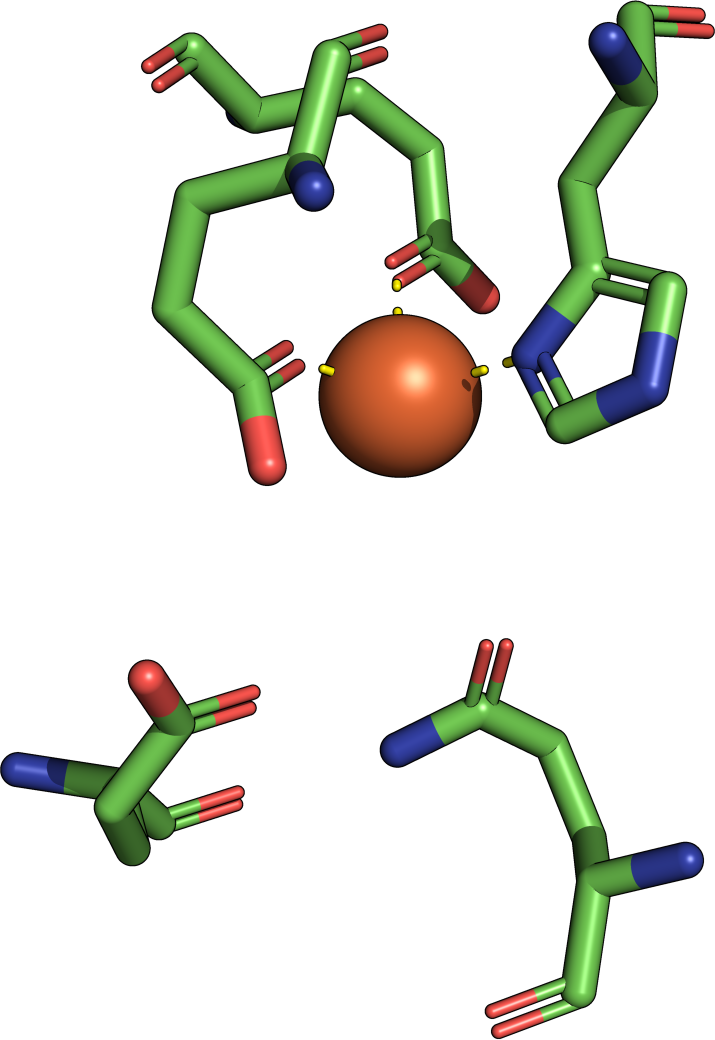
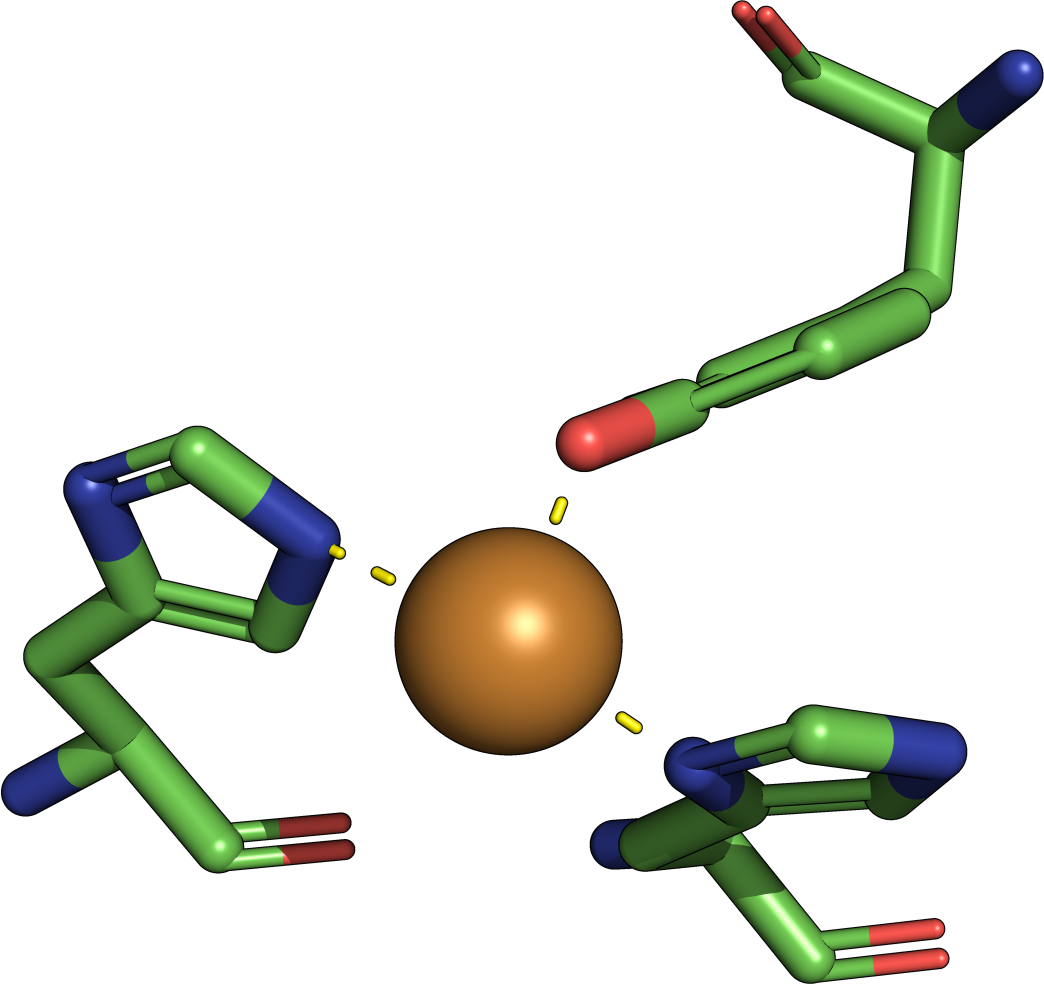
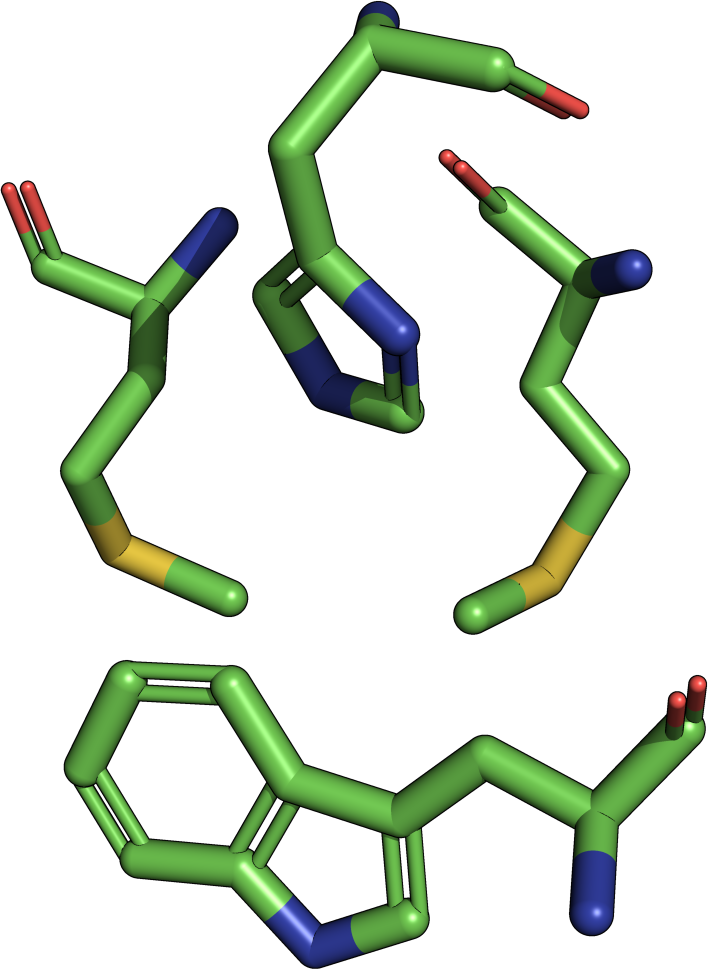
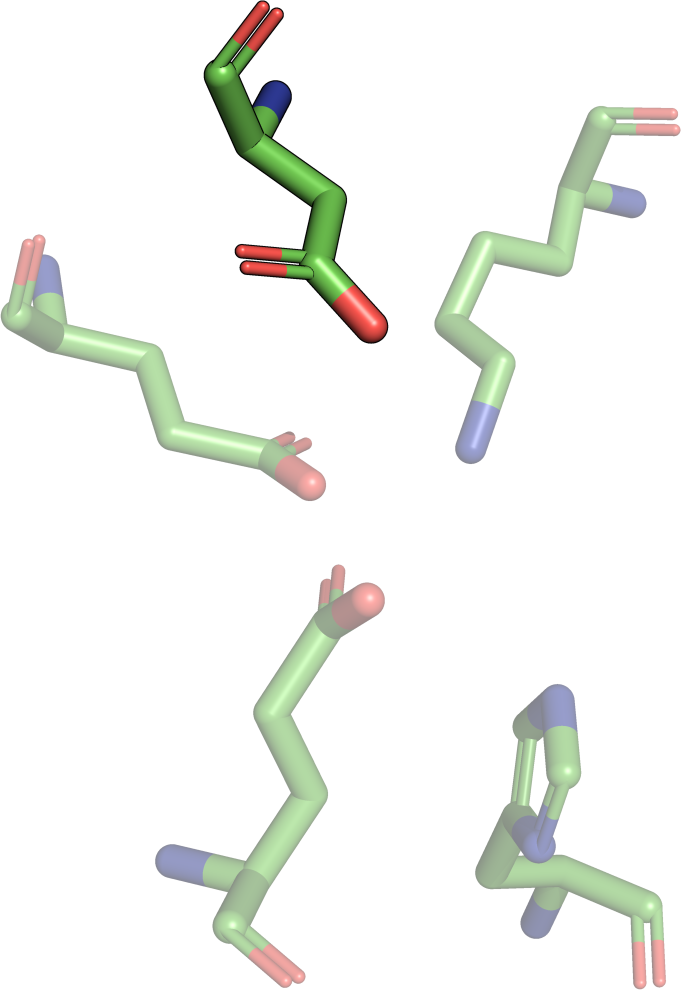
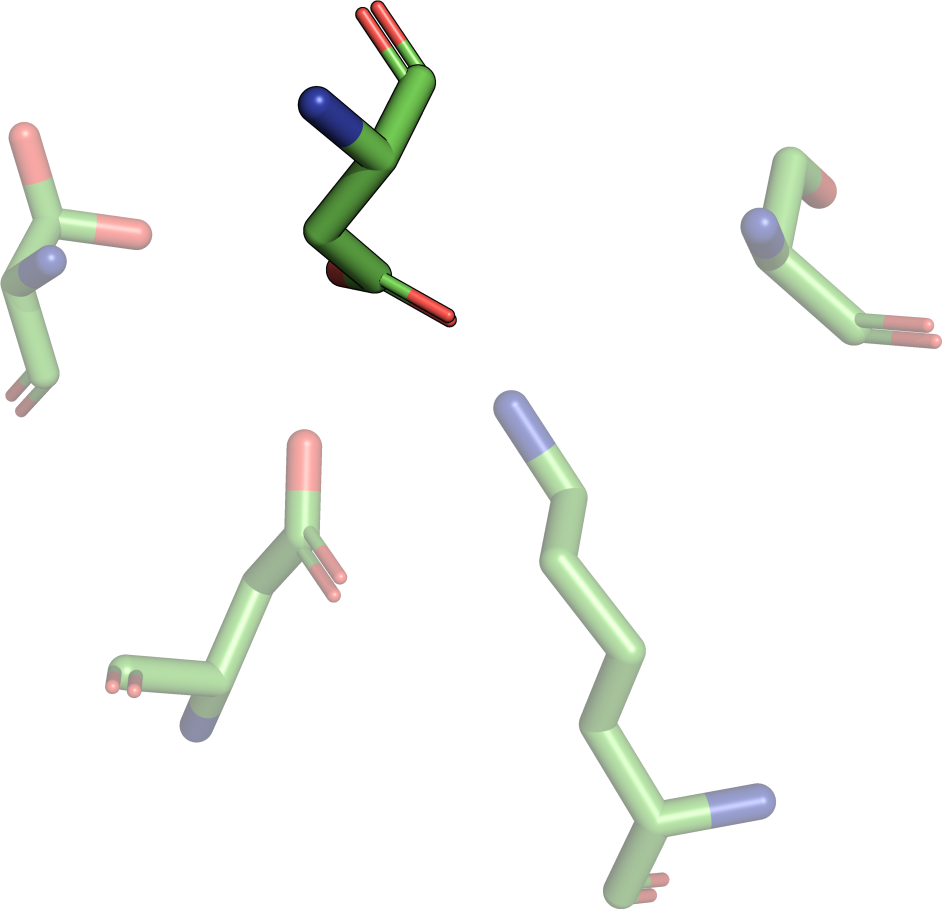
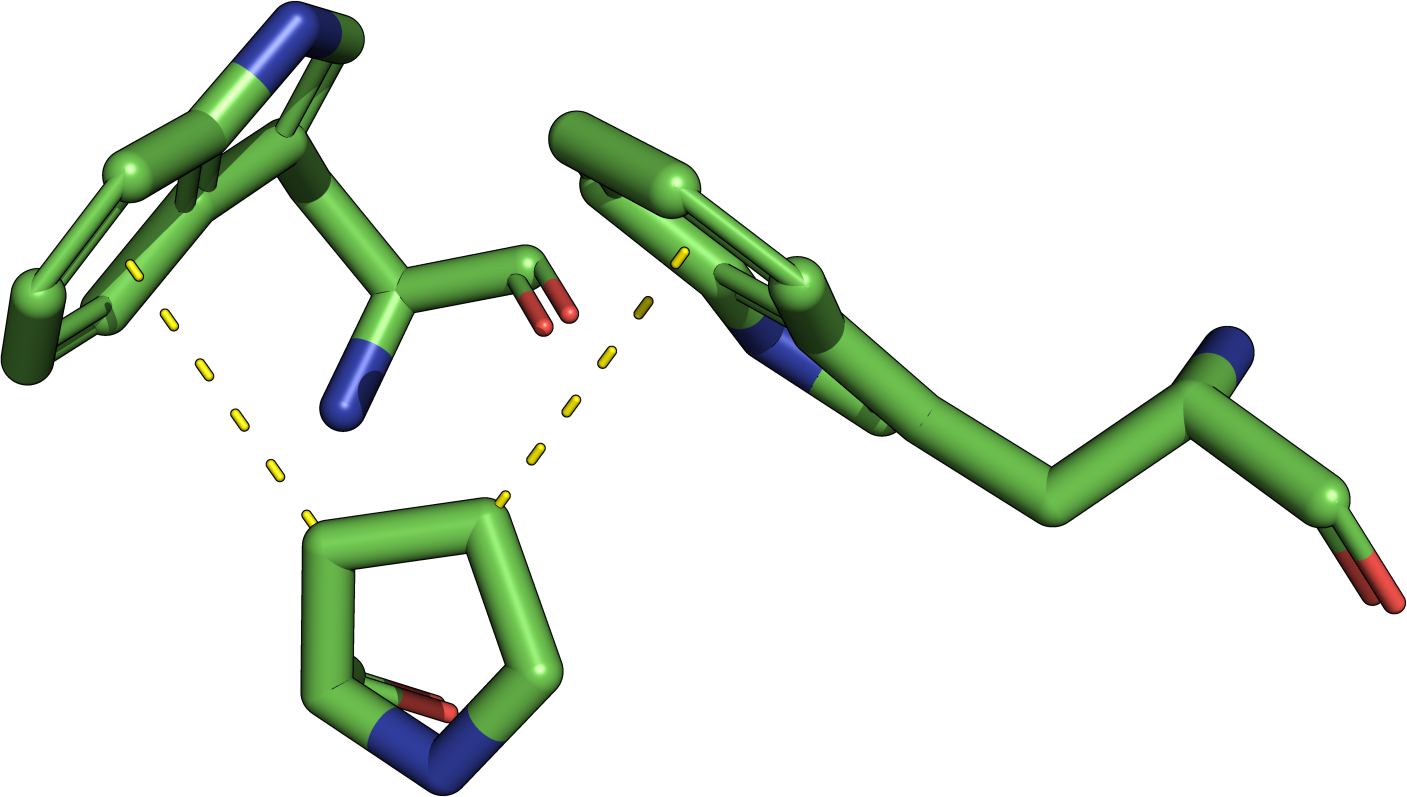
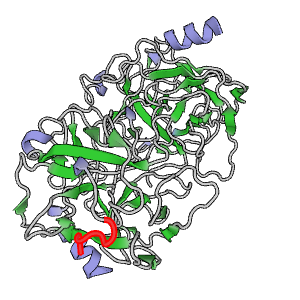
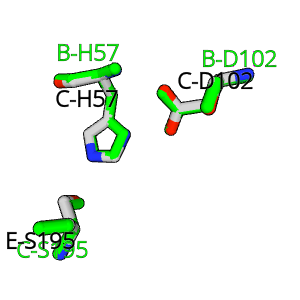
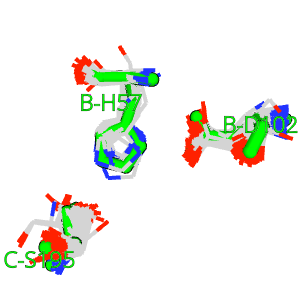
The theory behind the Fit3D algorithm was recently published in Journal of Computational Biology. Check out the paper to learn how our method works and to see how it performs in contrast to existing approaches.
| ||
| Kaiser, F., Eisold, A., Labudde, D. (2015) A novel algorithm for enhanced structural motif matching in proteins, J. Comput. Biol., 22, 698-713. | ||
|
| ||
| Kaiser, F., Eisold, A., Bittrich, S., Labudde, D. (2016) Fit3D: a web application for highly accurate screening of spatial residue patterns in protein structure data, Bioinformatics, 32-5, 792–794. | ||
|