

General
Proteins are important bio-molecules in biological systems and activities.
Knowledge of the protein structure gives us insight into function of the protein and its dynamics.
On the other hand, protein structure comparison is a fundamental task in structure biology.
The number of protein structures has grown rapidly over the last decade.
There is a need for new techniques which can rapidly compare protein structures with high performance and accuracy.
Protein structure comparison is crucial for understanding protein evolution, architecture and function.
A lot of tools and methods in the field of bioinformatics and structure biology are based on structure and/or sequence comparison.
This database and toolbox offers novel approaches based on so called energy profiles for comparison and prediction of globular and membrane protein structures.
Those profiles are calculated using a coarse-grained energy model.
Based on the frequency of residue contacts in known protein structures, the potential for pairwise residue-residue-interactions can been calculated.
An energy profile is a schematic plot of the interaction energy of each residue as a function of the residue position in the structure over the sequence.
As an abstraction of protein sequence and structure information, each energy profile is a protein specific fingerprint representation.
In the case of alpha-helical membrane proteins, 370 non-redundant polypeptide chains were extracted from the PDBTM database and their energy profiles were stored in the eProS database.
We provide two different potentials based on statistical analysis of the two protein structure sets separately.
With the help of our database and the involved tools it is possible to evaluate, compare and predict protein structures based on different input data.
The ePros database offers the opportunity to search for similar energy profiles in an internal database of almost 80,000 pre-calculated energy profiles.
Energy profiles of wild type and mutated proteins can be compared easily with the tools presented on these webpages.
Furthermore, the toolbox allows the calculation of an energy profile by almost any known protein structure.
Additionally, an energy profile can also be predicted from amino acid sequence.
These energy profiles can be visualized and/or aligned to other energy profiles using an adapted Needleman-Wunsch- or Smith-Waterman algorithm.
Basically, the alignment algorithms are modified by using pairwise energy scorings for weighting energetic (mis)matches.
Detected similarities or energetic divergences give insight to structural and functional relations.
The user has the opportunity to download the resulting energy profile files and re-use them as toolbox-input for further investigations as well.
This ensures that the user can continue his work at any point in time.
With the help of our database and the specifically concerted tools, it is possible to evaluate, compare and predict protein structures as well as estimate functional relationships and dynamics.
Provided annotations from CATH, SCOP, PDB and Gene Ontology aid to understand these correspondences more efficiently.
Theory of energy profiles
Energy profiles are derived by coarse-grained amino acid interaction models based on $ C_{\alpha} $ and $ C_{\beta} $ atom coordinate information extracted from known protein structures.
The propensity of an amino acid residue to interact with other residues (and adding information of the residue localization with respect to the membrane lipid bilayer in the case of the membrane proteins) is investigated.
In general, the energy of any protein is given by equation \eqref{eq:definition}, where the parameters $ n_{a_i}^{in} $ and $ n_{a_i}^{out} $ are equal to the number of observing the amino acid $ a_{i} $ being buried in the structure or exposed to the solvent at the protein surface, respectively.
These parameters are derived from known globular and membrane protein structures.
In our coarse-grained energy model, the interaction energy $ e_{a_{i},a_{j}} $ between two amino acids $ a_{i} $ and $ a_{j} $ is equal to the summation of $ e_{a_{i}} $ and $ e_{a_{j}} $ as depicted in equation \eqref{eq:single}.
Then the computation of the total energy $ E_{a_{i}} $ of amino acid residue $ a_{i} $ can be performed as given in equation \eqref{eq:total}.
By iterating over all amino acids in a protein structure, the total energy for each amino acid can be determined.
The sequence of all total energies corresponds to the energy profile.
In our work, we have shown that coarse-grained energies can be applied to investigate protein structure stability and functionality.
It is demonstrated that energy profile similarities point to common sequential, structural and functional characteristics.
Thus, changes of single energy values or changes in energy profile progression can be used for analyzing mutated characteristics of the proteins of interest.
\begin{eqnarray} e_{a_i} & \propto & -\ln{\left({n_{a_i}^{in} \over n_{a_i}^{out}}\right)} \label{eq:definition} \\ e_{a_{i},a_{j}} & = & \left( e_{a_{i}} + e_{a_{j}} \right) \label{eq:single} \\ E_{a_{i}} & = & \sum_{< i, j >}{e_{a_{i},a_{j}}} \label{eq:total} \end{eqnarray}
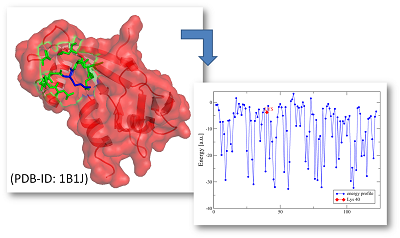
In all applications the energy profiles are visualised by a scheme of energy quantiles. The quantiles are derived from the distributions of energy values of all protein structures in our globular and alpha-helical transmembrane protein energy profile database. The coloring scheme is given as follows: blue - very low energy (very stable), green - low energy (stable), yellow - ambivalent energy (intermediate stability) and red - very high energy (instable).
Alignment of energy profiles
The alignment of energy profiles is based on dynamic programming and an adapted Needleman-Wunsch/Smith-Waterman algorithm (with cost schemes optimized for energy profile comparison).
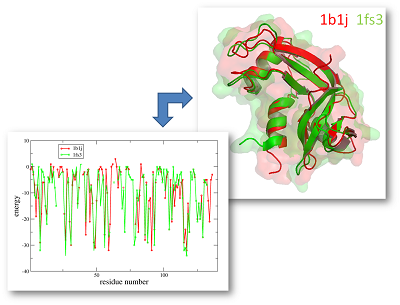

For this alignment of 1B1J and 1FS3 we would receive the following values:
sequence identity: 30.3%
sequence similarity: 47.0%
DaliLite zScore: 15.3
eAlign dScore: 1.4
The dScore (distance score) is calculated by a permutation based alignment evaluation approach leading to values between 0 (exact match) and 5 (no detectable correspondences).
Thus, the received dScore of 1.4 indicates significant energy profile similarity and alignment significance.
Prediction of energy profiles
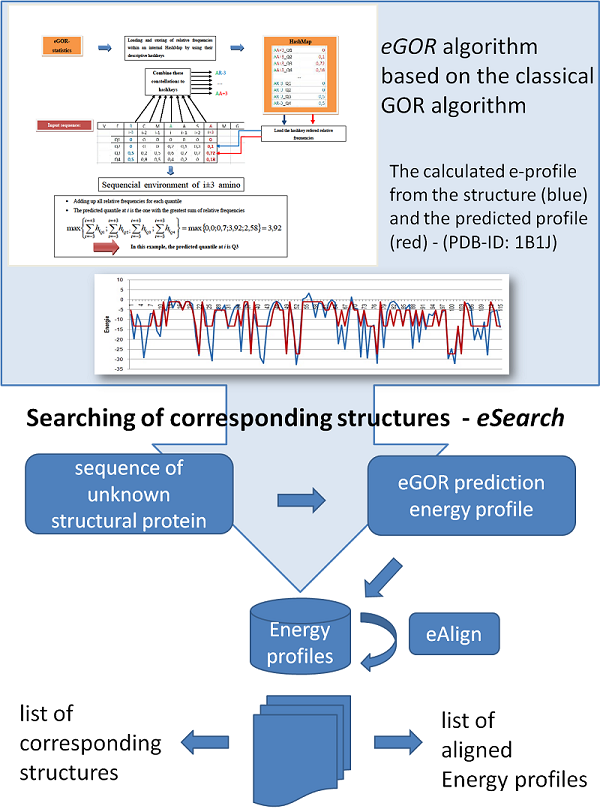
References
F. Heinke and D. Labudde. Membrane protein stability analyses by means of protein energy profiles in case of nephrogenic diabetes insipidus.
Comput Math Methods Med, 2012:790281, February 2012.
F. Heinke, A. Tuukkanen, and D. Labudde. Analysis of Membrane Protein Stability in Diabetes insipidus.
InTech, 2011, Edited by Kyuzi Kamoi,ISBN: 978-953-307-367-5,DOI: 10.5772/22258
F. Heinke and D. Labudde. Predicting functionality of the non-expressed putative human OHCU decarboxylase by means of novel protein energy profile-based methods.
Conference proceedings of 13. Nachwuchswissenschaftlerkonferenz, April 2012.